
Dr. Matthew J. Young received his B.Sc. and Ph.D. degrees from the University of Manitoba. Dr. Young was a Postdoctoral Fellow at the National Institute of Environmental Health Sciences (NIEHS) and joined the SIUSM faculty in 2015.
Office: Neckers Rm 211
Office phone: (618) 453-6437
Laboratory: Neckers Rm 206
Lab Phone: (618) 453-1132
email: matthew.young@siu.edu (preferred)
(or myoung84@siumed.edu)
Surprisingly, mtDNA mutations are common in the population and it has been estimated that one in 200 healthy humans harbors a pathogenic mtDNA mutation that potentially causes disease in the offspring of female carriers (Elliott et al. AJHG 2008). Other researchers have estimated the incidence of mitochondrial disease to be about one in 5,000 (Wallace and Chalkia. CSH Perspect Biol 2013) and mitochondrial dysfunction is at the root of genetic disorders that affect both older adults and young children. Diseases associated with mitochondrial dysfunction include Parkinson's, Huntington's, Alzheimer's, ALS, epilepsy, and cancer.
Human mtDNA is maternally inherited and occurs as a double-stranded negatively supercoiled circular genome of 16,569 base pairs (bp) that encodes 37 genes required for energy production. The remaining mitochondrial genome includes greater than 1,000 nuclear genes. Mutations in a class of these genes are associated with Mendelian disorders. When cells in the body lose their mtDNA, or mtDNA becomes mutated, mitochondria become dysfunctional and mitochondrial disease ensues.
Human mtDNA is a multicopy genome occurring at about 1,000 copies per cell and these molecules are maintained by the replicative mtDNA polymerase, pol γ. Pol γ is a trimer composed of subunits encoded by two nuclear genes, the POLG gene encoding the monomeric p140 catalytic subunit and the POLG2 gene encoding the p55 homodimeric processivity subunit. Mitochondrial diseases, including those linked to POLG and POLG2 mutations, are devastating incurable disorders. Pol γ functions in conjunction with additional DNA polymerases and other mtDNA maintenance machinery to replicate and repair the mtDNA genome (Fig. 1). In addition to mutations in pol γ genes, disease mutations exist in mtDNA maintenance machinery genes including the Twinkle mtDNA helicase, the helicase/nuclease DNA2, the 5'-3' MGME1 exonuclease, the RNaseH1 enzyme required to remove RNA primers, and the multifunctional mtDNA packaging factor TFAM (Transcription Factor A Mitochondria).
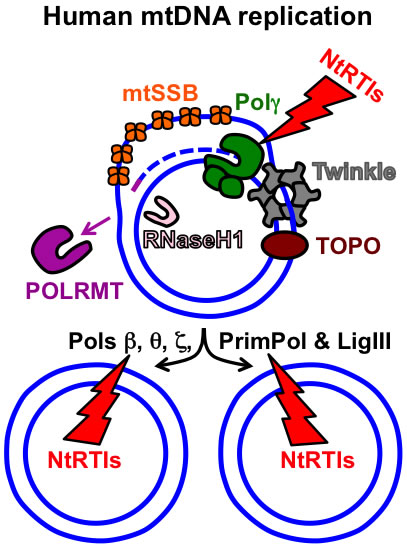
Figure 1. Key components of the human mtDNA maintenance machinery. The blue circles represent the double-stranded circular mtDNA genome. The small purple line represents an RNA primer while the blue dashed line represents newly synthesized mtDNA. Pol γ, the replicative mtDNA polymerase gamma; TOPO, topoisomerase; Twinkle, Twinkle mtDNA helicase; POLRMT, mitochondrial RNA polymerase and primase; RNaseH1, Ribonuclease H1; mtSSB, mitochondrial single-stranded DNA binding protein. DNA polymerase beta, theta, zeta, and the DNA primase and translesion DNA polymerase are represented by Pols β, θ, ζ, and PrimPol respectively. The additional polymerases likely assist Pol γ with overcoming mtDNA damage. The NtRTIs lightning bolt represents nucleotide reverse transcriptase inhibitors blocking DNA polymerases. See https://www.frontiersin.org/articles/10.3389/fmolb.2017.00074/full for further details on additional factors involved with mtDNA replication and repair.
The Young laboratory uses a variety of human cell culture, bioenergetic, molecular biology, and fluorescence microscopy techniques to study human mitochondrial function. In addition to genetic disease mutations that disrupt mtDNA replication, evidence supports that mitochondria are targets of toxicants that disrupt mtDNA replication. MtDNA depletion can be a side effect in human immunodeficiency virus (HIV)-infected subjects treated with nucleoside reverse transcriptase inhibitors (NRTIs) and mitochondrial toxicity from NRTIs mimics phenotypes of mitochondrial disease such as mitochondrial myopathy (M. J. Young, 2017). Also, cell culture studies have demonstrated that additional stressors can cause mtDNA degradation or mtDNA depletion.
The mission of our laboratory is to understand the molecular mechanisms that cause mitochondrial dysfunction in human health and disease. The aims of this research include determining how genetic mitochondrial disease mutations, as well as toxicants and drugs, disrupt mitochondrial function. We are currently investigating off-target effects of drugs on mitochondrial function using a cell line model of liver cells, HepaRG. Lastly, we are designing and characterizing cell line models of mitochondrial disease.
Wheeler JH, Young CKJ, and Young MJ. Analysis of human mitochondrial DNA content by Southern blot and non-radioactive probe hybridization. Curr Protoc Toxicol. Accepted March 25, 2019.
Young CKJ and Young MJ. Comparison of HepaRG cells following growth in proliferative and differentiated culture conditions reveals distinct bioenergetic profiles. Cell Cycle, 1-24. 2019. PMID: 30755072 (https://www.tandfonline.com/doi/full/10.1080/15384101.2019.1578133).
Young MJ. Off-Target Effects of Drugs that Disrupt Human Mitochondrial DNA Maintenance. Front Mol Biosci, 4, 74. 2017. PMC5702650 (https://www.frontiersin.org/articles/10.3389/fmolb.2017.00074/full).
Young MJ and Copeland WC. Human mitochondrial DNA replication machinery and disease. Curr Opin Genet Dev. Apr 8;38:52-62. 2016. PMID: 27065468. *Article highlighted on the cover.
Al-Bagdadi F, Young MJ, Geaghan JP, Yao S, Barona HM, Martinez-Ceballos E, & Yoshimura M. Observation on the ultrastructure morphology of HeLa cells treated with ethanol: Statistical analysis. Ultrastruct Pathol, 40(6), 324-332. 2016. PMID 27680498.
Young MJ, Humble MM, DeBalsi KL, Sun KY, and Copeland WC. POLG2 disease variants: analyses reveal a dominant negative heterodimer, altered mitochondrial localization and impaired respiratory capacity. Hum Mol Genet. Sept15;24(18), 5184-5197. 2015. PMID: 26123486.
Craig K+, Young MJ+, Blakely EL, Longley MJ, Turnbull DM, Copeland WC, and Taylor RW. A p.R369G POLG2 mutation associated with adPEO and multiple mtDNA deletions causes decreased affinity between polymerase γ subunits. Mitochondrion. Mar:12(2):313-9. 2012. PMID: 22155748. +MJY and CK contributed equally to this work.
Bay DC, Hafez M, Young MJ, Court DA. Phylogenetic and coevolutionary analysis of the β-barrel protein family comprised of mitochondrial porin (VDAC) and Tom40. Biochim Biophys Acta. Jun; 1818(6):1502-19. Dec 4. 2012. PMID: 22178864.
Young MJ, Longley MJ, Li FY, Kasiviswanathan R, Wong LJ, and Copeland WC. Biochemical analysis of human POLG2 variants associated with mitochondrial disease. Hum Mol Genet. Aug 1:20(15):3052-66. Epub May 9. 2011. PMID: 21555342. *Article highlighted on the cover.
Kasiviswanathan R, Longley MJ, Young MJ, Copeland WC. Purification and functional characterization of human mitochondrial DNA polymerase gamma harboring disease mutations. Methods. Aug;51(4):379-84. 2010. PMID: 20176107.
Young MJ and Court DA. Effects of the S288c genetic background and common auxotrophic markers on mitochondrial DNA function in Saccharomyces cerevisiae. Yeast. Dec; 25(12): 903-12. 2008. PMID: 19160453.
Young MJ+, Bay DC+, Hausner G, and Court DA. The evolutionary history of mitochondrial porins. BMC Evol Biol Feb 28;7:31. 2007. PMID: 17328803. +MJY and DCB contributed equally to this work.
Young MJ, Theriault SS, Li M, and Court DA. The carboxyl-terminal extension on fungal mitochondrial DNA polymerases: identification of a critical region of the enzyme from Saccharomyces cerevisiae. Yeast. Jan 30; 23(2):101-16. 2006. PMID: 16491467.
Young MJ and Court DA. Quick Measurement of Glucose Concentration in Saccharomyces cerevisiae Cultures. The Genetics Society of Canada Bulletin. 35(4): 109-110. 2004. Bulletin.
Dr. Matt Young, PI
Ms. Carolyn Young, carolyn.young@siu.edu, Researcher II
Ms. Grace Forrest, Researcher I
Mr. Joel Wheeler, M.S. Student
Ms. Isabel Yunker, Undergraduate Student Worker
Mr. Michael Lay, Undergraduate Student
https://www.researchgate.net/lab/The-Young-Lab-at-SIUSM-Matthew-J-Young
http://atlasofscience.org/mitochondrial-polg2-disease/
https://www.ninds.nih.gov/Disorders/All-Disorders/Mitochondrial-Myopathy-Information-Page
![]() Biochemistry and Molecular
Biology Home Page
Biochemistry and Molecular
Biology Home Page